NMO是一种以严重的视神经炎和长节段横贯性脊髓炎(LETM)发作为特征的中枢神经系统炎症性疾病。过去的几十年里,我们对NMO的认识已取得巨大的进步。这主要得益于疾病特异性的自身抗体,NMO-IgG,以及随后其主要的自身靶抗原,水通道蛋白4(AQP-4)抗体的发现,从而使得NMO与MS区别开来,成为一个独立的疾病。
目前的诊断标准(指2006年的诊断标准)中,NMO的诊断依然需要出现视神经炎和脊髓炎。不过,AQP-4抗体的发现使得我们认识到除了经典的NMO外,还存在一组更为多样的临床表型,即所谓的“视神经脊髓炎谱系疾病(NMOSD)”。NMOSD不仅包含了血清AQP4抗体阳性的伴有特定脑部异常的限制型NMO(limited forms of NMO)或经典型NMO,还包括伴有其他自身免疫性疾病(例如SLE,干燥综合征等)的血清AQP4抗体阳性的患者。这种情况下,MRI在NMOSD与其他中枢神经系统炎症性疾病尤其是MS的鉴别中扮演着越来越重要的角色。因为治疗各异,因此将上述疾病区分开来很关键。此外,当前不断发展的MRI技术可帮助发现更多的特异性标志物,有助于阐明NMOSD潜在的致病机制。
NMO患者的早期头颅MRI研究发现了一些无法解释的临床无症状的、非特异性的白质病变。AQP4-IgG检测的出现更新了我们的认识,实际上很大一部分NMOSD患者存在脑部MRI的异常,常位于AQP4高表达区域。但AQP4表达不高的区域也可有脑部异常。虽然T2WI/FLAIR上非特异性的小点状或斑片状高信号是NMOSD最常见的影像学表现,但NMOSD某些病灶仍具有部位或形态学上的特征。
在AQP4抗体发现前,报道的脑部MR异常仅见于13-46%的NMO患者。但是,当不考虑头颅磁共振标准时,头颅MRI异常可达50-85%(参考修订版2006年NMO诊断标准)和51-89%(NMOSD血清抗体阳性患者)。此外,据报道,NMOSD发病初期即有头颅MRI异常的患者约43-70%。不同文献报道的头颅MRI异常的比例差异很大,一种可能解释是,随着疾病进展,头颅MRI异常出现的概率越来越高。在一组包含88例血清抗体阳性的儿童报道中,行头颅磁共振检查的患儿中有68%存在头颅异常,其中病灶在第三脑室周围区域(间脑)、第四脑室周围区域(脑干)、幕上和幕下的白质、中脑和小脑等部位更为明显。这与观测到45-55%的NMOSD患儿有发作性脑部症状这一现象是吻合的,临床表现包括眼肌麻痹、顽固性呕吐和呃逆、意识状态改变、严重的行为学改变、嗜睡、共济失调和癫痫发作。
NMOSD的头颅MRI表现分类
1环绕脑室系统的室管膜周病变
(1)环绕第三脑室和导水管的间脑病变:环绕第三脑室和导水管的间脑病变部位包括丘脑、下丘脑、中脑前界,在NMOSD中已有报道(图1A)。这些病变通常是无症状的,但是有些患者可能会出现抗利尿激素分泌异常、嗜睡发作、低体温、低血压、睡眠过度、肥胖、甲状腺功能减退、高泌乳素血症、继发性闭经、溢乳和行为改变等症状。
(2)邻近四脑室的脑干背侧病变:邻近四脑室的脑干背侧包括极后区(area postrema)和孤束核的病变是NMOSD患者最为特异性的头颅MRI表现之一。见于7-46%的NMOSD患者,并且与顽固性呃逆、恶心、呕吐有高度相关性。这一区域为呕吐反射的中枢,有着较为疏松的血脑屏障,因而更容易受到AQP4-IgG的攻击。MRI和临床证据均提示极后区是NMOSD易受攻击的重要区域,后续研究发现该区域是血液循环中的IgG进入中枢神经系统的重要通道。40%的NMO患者这一区域有病理改变,但是没有明确的神经元、轴索或髓鞘缺失。延髓病变常与颈髓病变相连续,多呈线形(图1B.b)。这些病灶常与疾病的第一症状或者预示急性恶化有关。脑干病变可出现各种不同的临床表现,例如眼球震颤、构音障碍、吞咽困难、共济失调、眼肌麻痹等。
(3)环绕侧脑室的室管膜周病变:胼胝体病变见于12-40%的NMOSD患者。因为NMO和MS患者都常出现胼胝体病变,所以这一部位并不能作为NMO和MS的特异鉴别点。但是,MS的胼胝体病变常常是不连续的、卵圆形、垂直于侧脑室,多累及胼胝体下部(图2A)。而NMOSD病变位置与侧脑室很接近,紧贴室管膜内层(图1C.a)。NMOSD急性期的胼胝体病变常有明显水肿,且为多形性,形成“大理石样图案(marbled pattern)”,有时累及胼胝体压部全层,出现独特的“拱桥形图案(arch bridge pattern)”(图1C.b和C.c)。有时,胼胝体病变延续至大脑半球,形成广泛、融合的白质病变。在NMOSD的慢性期,胼胝体病变可逐渐缩小、信号减弱,甚至可以消失;但是,囊变和胼胝体萎缩都曾有过报道。某些临床表现,如认知功能和运动协调能力障碍等,可能与胼胝体受损有关,但证据并不是很充分。
2大脑半球的白质病变
广泛、融合的大脑半球白质病变常呈瘤样(最大半径可以>3cm),或者沿白质纤维走行呈长纺锤状或放射状(图1D)。通常无占位效应。ADC上病灶弥散系数增高,提示可能为急性炎症相关的血管源性水肿(图1D.c),可能与可逆性后部脑病综合征(PRES)或Balo病混淆。相较于AQP4抗体阴性的患者,这些广泛的病变在AQP4抗体阳性的患者中更常见。疾病慢性期,这些大病灶趋向于缩小甚至消失,但有一些患者可出现囊样或空洞型改变。上述病变根据其累及区域的不同可引起各种症状,如偏瘫、脑病、视野缺损等。大片融合的大脑半球白质病变在NMOSD患儿中比较常见。伴随着灶周水肿和不同程度占位效应的肿瘤样病灶可类似急性播散性脑脊髓炎(ADEM)或中枢神经系统恶性肿瘤。
3病变累及皮质脊髓束
皮质脊髓束受累可为单侧或双侧,病灶可能从大脑半球深部的白质通过内囊后肢延伸至中脑的大脑脚或者脑桥(图1E)。这些病灶连续,常为长节段,沿锥体束分布(图1E.c)。一些关于NMOSD患者的队列研究报道,约23-44%的患者存在皮质脊髓束的病灶,在其他研究中也偶有发现。有意思的是,和脑室周围不同,皮质脊髓束并不是AQP4高表达的区域,因此目前尚不清楚NMOSD患者这一区域常累及的原因。
4非特异性病灶
T2WI/FLAIR上,皮质下区或深部白质区常可观察到非特异性的小点状(<3mm)或片状高信号,这是NMOSD最常见的头颅MRI异常(35-84%),通常无临床症状。
5强化病灶
先前的一些研究报道9-36%的NMOSD患者头颅MRI可出现强化病灶,但具体比例尚不清楚。大多数病灶呈边界不清、模糊的多发片状强化,即所谓的“云雾状强化”(图1F.a)。这种云雾状的强化模式有助于与边界清楚的卵圆形或环状/开环状病灶(图2)鉴别,后者为MS较为典型的特征。侧脑室室管膜表面的线状强化(铅笔样病灶)也有报道(图1F.b)。边界清楚的结节样强化或脑膜强化也可见于NMOSD患者,但罕见。
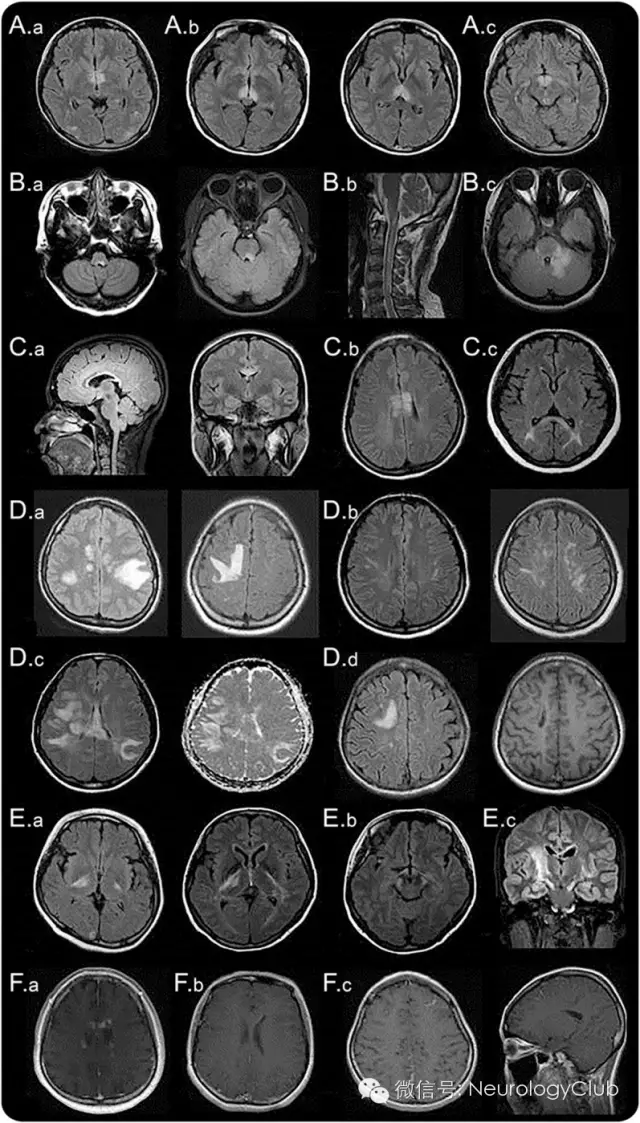
(Aa:环绕第三脑室和导水管的间脑病变;Ab:丘脑、下丘脑受累;Ac:中脑前界;Ba:邻近四脑室的脑干背侧病变;Bb:与颈髓病变相连续的延髓线状病变;Bc:肿胀弥漫的脑干背侧病变,累及小脑脚;Ca:邻近侧脑室、紧贴室管膜内层的胼胝体病变;Cb:胼胝体病变呈“大理石样图案” ;Cc:胼胝体病变呈“拱桥形图案”;Da:肿瘤样大脑半球白质病变;Db:长纺锤状病灶;Dc:ADC上病灶弥散系数增高,提示血管源性水肿;Dd:慢性期大脑半球病灶囊性变;Ea:内囊后肢皮质脊髓束病变;Eb:中脑大脑脚病变;Ec:沿锥体束的长节段病变;Fa:云雾状强化;Fb:侧脑室室管膜表面线状强化;Fc:脑膜强化)
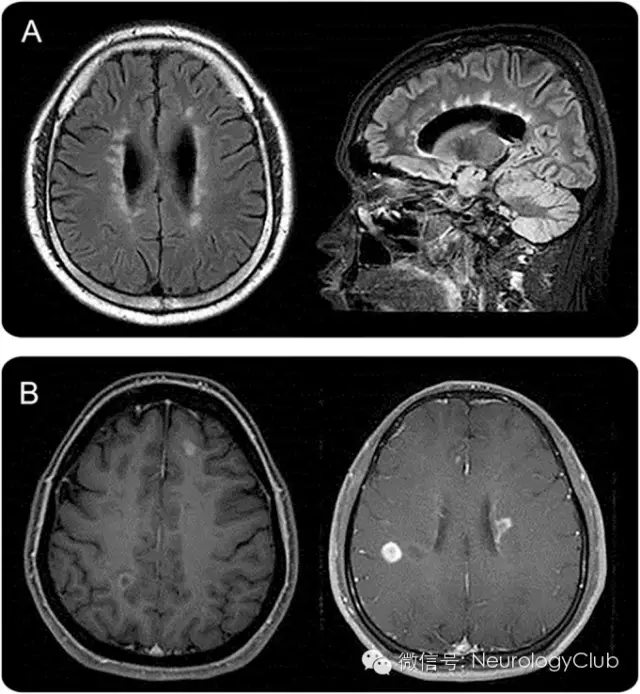
(A:侧脑室与胼胝体可见不连续的,卵圆形的,垂直于侧脑室的病灶;B:病灶呈卵圆形或开环强化,边界清楚)
NMOSD的视神经MRI表现
目前已有报道认为,在急性视神经炎期,T2WI和T1WI增强序列可见非特异性的视神经鞘增厚和视神经高信号。但因为MS患者视神经炎也可有类似改变,所以这一特点无法作为NMOSD的诊断支持点。现阶段的研究主要着眼于MS和NMOSD中视神经损伤磁共振的不同特点。NMOSD患者中,病变多累及包括视交叉在内的视神经后部,常同时有双侧视神经受累。因此,在临床实践中,当出现视神经长节段炎症,特别是同时存在双侧受累、病变向后延伸累及视交叉时,应考虑NMOSD的可能。
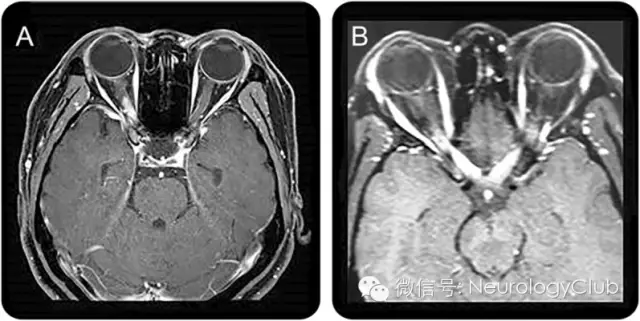
(A:右侧视神经后部强化病灶;B:双侧视神经后部/视交叉弥漫强化病灶)
NMOSD的脊髓MRI表现
NMOSD患者脊髓的炎性病变以T1WI低信号,T2WI高信号为特征。研究认为,相对于腰髓和下胸髓,这些病变更多见于颈髓和上胸髓,且倾向累及灰质。脊髓中的AQP4在灰质以及中央管的室管膜周围的胶质细胞中比较丰富,只有少部分位于脊髓白质中。
NMO最特征性的表现为长节段横贯性脊髓炎(LETM),即脊髓病变长度大于3个连续的脊髓节段,脊髓MRI上可见病灶主要累及中心灰质区域(图4)。但是,并不是所有的LETM都是NMOSD,目前有几组针对LETM患者的研究显示,AQP4抗体阳性组和阴性组在人口学和临床特点上均有明显差异。LETM对于诊断NMO的特异性在儿童患者中要低于成人。LETM常见于罹患急性播散性脑脊髓炎的患儿,也可在17%的MS患者中出现,还可在67-88%的单相病程的横贯型脊髓炎患儿中观察到。因此,当患者表现为LETM时,需要考虑除了NMOSD以外的其他疾病的可能。
NMOSD随访过程中的脊髓病变:NMOSD病程中LETM的MRI改变已被观察到,MRI资料显示,LETM在疾病恢复期或者应用大剂量糖皮质激素治疗后,可能发展为多个短的病灶。此外,复发性脊髓炎可导致脊髓萎缩,可能引起神经功能障碍。因此,定期行MRI检查对于诊断LETM有着重要意义。
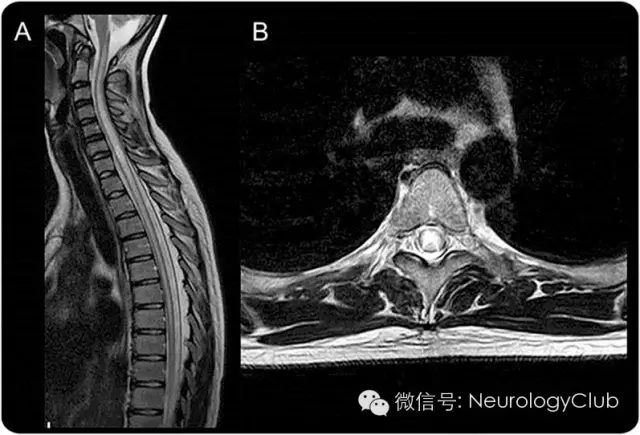
(A:累及胸髓的长节段病变;B:灰质选择性受累[H形脊髓病变])
NMOSD和MS的影像学比较
临床实践中,NMO的主要鉴别诊断为MS,特别是当病变局限于视神经和脊髓时。这两种疾病的鉴别很重要,早期及准确诊断意义重大,因为治疗和预后不尽相同,有些MS治疗方案可加重NMO。分析比较MS和NMOSD之间的特点有助于对两者不同病理过程的理解。
对于NMOSD的患者,我们有特异性的生物标志物(血清AQP4抗体)可以用于筛选,但是MS没有相应的特异性生物标志物。NMO和MS的对比研究常应用不同的选择标准,特别是是否限制AQP4抗体阳性作为NMO的准入标准,这将会影响到最终的结果。一些有争议的数据一部分可以用AQP4抗体的检测手段不同来解释,因为不同方法的敏感性不同并且会因处在疾病随访的不同阶段而产生差异。
如上文所述,NMO最重要的影像学特征是LETM,但是有些患者仅表现为位于脊髓中央的短节段脊髓炎。其他NMOSD和MS的脊髓病变的不同MRI特点如下表所示。
2006年NMO诊断标准中包含了发病初期头颅MRI不符合MS影像学诊断(Paty标准)作为NMO的支持诊断。但是,MS样病灶也可见于10-12.5%的NMO病例中,并且5-42%的NMO患者满足Barkhof标准。近期的一篇报道显示,分别有13%和9%的NMOSD患者发病初期的脑部病变满足Barkhof标准和欧洲MS磁共振诊断标准。AQP4抗体阳性的患者和MS患者之间尚不存在有统计学差异的病灶分布。但是,MS头颅MRI的一些典型特征具有一定的敏感性和特异性,例如侧脑室和颞叶下病灶的出现,Dawson手指征或S形U形纤维病灶等,倾向于诊断MS。通过皮层病变敏感性较高的影像手段已经证实NMO患者不出现皮层病灶,但皮层病变见于多数MS患者。在高场强MRI下,环绕中心小静脉的特征性MS脑部病灶可见于>80%的患者中。而在NMO患者中,出现概率要小得多,约9-35%,可能提示两种疾病不同的致病机制。
两种疾病中静止性病灶出现的概率也有差别。相比MS,NMOSD患者更少出现临床静止性MRI病灶。然而,新的静止性病灶确实可在少部分NMOSD患者中出现。此外,多数研究发现,通过非常规影像学手段,例如弥散张量成像,可观察到MS患者中非病变组织损害,而在NMOSD患者中不常见,除了连接病变上下行传导束。总的来说,上述发现与NMO临床过程符合,相对于MS来说,NMO可能是一种病变依赖性疾病,可有复发但不伴有广泛的神经退行性病理过程,因此疾病没有进展期。
NMO和MS之间的不同点可能是由于NMO是中枢神经系统特异性的抗体介导的针对星形胶质细胞的病理过程,而非针对髓鞘的T细胞为主的炎症反应所致。支持这种可能性的是,AQP4抗体阳性的颈髓病变患者中肌醇(一种星形胶质细胞的功能标志物)减少,而MS患者中无此改变;与对照组相比,MS患者中N-乙酰天门冬氨酸(NAA)(一种髓鞘和特异性神经丝损伤标志物)减少,而AQP4抗体阳性患者也有减少,但无统计学意义。
NMOSD和MS间重要的影像对比已总结在下表中。因为关于NMO长期系统的影像随访还未开展,其与MS的不同之处还需要进一步研究证实。
表 NMOSD和MS的MRI特点比较
| NMOSD | MS |
脊髓 | 长节段弥漫性损伤 | 短,常为多发病灶 |
| 中心/灰质受累 | 外周/非对称/常常在后部 |
| 急性病变T1常呈低信号 | T1低信号罕见 |
视神经 | 长段/后部-视交叉病变 | 病变较短 |
脑 | 环绕脑室系统的室管膜周围病变 | Dawson手指征(垂直于侧脑室)/S形U型纤维病变,侧脑室和颞叶下部病变 |
| 大脑半球肿瘤样病灶 | 皮层病变 |
| 皮质脊髓束受累 | 静脉周围病变 |
| 云雾状强化病灶 | 卵圆形或环形/开环形强化 |
其他 | 局限于病灶传导束和相关皮层的正常外观组织受累 | 特殊的磁共振成像可发现正常外观的白质损害 |
| MRS上病灶部位肌醇减少 | MRS上病变区域NAA减少 |
MRI异常对预后的提示作用
AQP4抗体被认为是一个预后标志物,阳性提示有较高的视神经炎和脊髓炎的复发风险。较多影像特点的出现可能提示脊髓的严重损伤,比如T1低信号伴水肿或空洞和萎缩,这些NMOSD患者往往预后差,出现难治性疼痛,且永久性残疾的风险高。此外,NMOSD患者上颈髓病变延伸至脑干者可出现呼吸衰竭的风险。
NMOSD患者急性发作期脑脊液中高水平的胶质纤维酸性蛋白(GFAP)往往与MRI脊髓病变长度和发作6月后扩展残疾状况评分量表分值有关。这种相关性可能意味着影像学表现与星形胶质细胞的损伤成比例,并且对预后有一定的提示作用。广泛出现的脑部病变可能提示随访期间更高的复发率和致残率。对于伴脑部病变的患者是否比不伴脑部病变的患者预后更差,还需要长期的随访来证实。就这点而言,目前尚无一个独立的MRI参数可以预测NMO的预后。
近来,髓鞘少突胶质细胞糖蛋白(MOG)抗体在一些符合NMOSD临床表现但AQP4抗体阴性的患者中被发现。比较AQP4抗体阳性和两种抗体均为阴性的患者,MOG抗体阳性但AQP抗体阴性的患者似乎发作次数更少,多为双侧视神经炎,脊髓炎位置更偏尾部,恢复更好。因此,MOG阳性患者与AQP4阳性患者相比,可能存在不同的致病机制,并且有更好的预后,但这仍然需要进一步的研究来证实。
总结展望:NMO诊断标准中的MRI特点
脑部MRI异常在NMOSD患者中很常见这一概念完全否定了先前认为颅脑MRI正常是NMO诊断的一个必要条件。本文复习总结了我们对于NMOSD影像表现的认识。但是NMOSD影像特点的敏感性和特异性都没有通过前瞻性的方法系统研究过,且这些影像学特征都无法确诊NMOSD或者作为NMOSD的诊断证据。因此,和其他中枢神经系统炎症性疾病一样,影像学表现可以拓宽诊断思路,这也是本综述想阐述的重要内容。实际上,比较实用的可以用来鉴别NMO和MS的主要还是一些比较传统的支持MS诊断的征象,如Dawson手指征。近期观察到的MOG抗体阳性患者的出现使得本病更为复杂,现认为,一些MOG抗体阳性患者(并非全部)可能属于NMO谱系疾病。
两者在临床表现、病因学以及影像学表现上的共同点以及差异,提示NMOSD可能不是一个同质性的疾病分类实体。另外,因为NMOSD可以与其他自身免疫性疾病共同出现,所以针对中枢神经系统的其他自身抗体如抗NMDA受体抗体等可以与AQP4抗体同时出现,多种中枢神经系统自身抗原的自身免疫反应可能参与了NMOSD的炎性病变的形成。本文总结的各种神经影像学表现反映了NMOSD的异质性。影像学技术的提高,包括高分辨率成像、特殊靶病灶的3D成像、计算机引导分析影像的定量比较,有助于更好的诊断、预后、治疗效果以及患者照护。目前正在进行的国际协作将提供更多,更典型的NMO/NMOSD患者,便于我们更好地研究脑部受累的概率,更全面地了解MRI异常在临床诊断和预后判断方面的价值。
fw译文,zyx审校
[参考文献]
Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, Klawiter EC, Sato DK, de Seze J, Wuerfel J, Banwell BL, Villoslada P, Saiz A, Fujihara K, Kim SH; Guthy-Jackson Charitable Foundation NMO International Clinical Consortium & Biorepository.MRI characteristics of neuromyelitis optica spectrum disorder: an international update.Neurology. 2015 Mar 17;84(11):1165-73.

神经病学俱乐部 微信号:NeurologyClub神经病学俱乐部,立足一线临床,服务神经科同行,助力神经病学天天向上