抗N-甲基-D-天冬氨酸受体(NMDAR)脑炎是一种抗NMDAR抗体介导的自身免疫性脑炎,主要表现为精神行为异常、意识障碍、癫痫发作、运动障碍、低通气、自主神经功能紊乱等,其诊断有赖于血清及脑脊液中检测到抗NMDAR抗体,半数以上患者头MRI多于海马、颞叶、额叶、胼胝体、大脑/小脑皮质、基底节、脑干等部位出现长T2、FLAIR高信号异常病灶。其中伴多发性硬化样脱髓鞘者较为少见,现将我们收治的1例报道如下。
患者男性,35岁,工人,因“反复发作复视、肢体抽搐、精神行为异常5年,发作性右下肢无力半年”于2015年7月12日入院。患者5年前(2010年7月)无明显诱因出现视物成双、视物模糊,后出现头痛、发热,体格检查见双眼水平眼震、双下肢病理征阳性,化验脑脊液白细胞20×10^6/L、白蛋白0.46 g/L,颅脑MRI平扫未见明显异常,诊断为“病毒性脑炎”,给予“抗病毒、激素”等治疗37 d后症状缓解出院。2011年7月患者出现发作性意识不清、四肢抽搐,行头颅MRI示“左侧侧脑室后角白质脱髓鞘改变”(图1A-C),诊断为“脱髓鞘病变”,给予甲泼尼龙冲击治疗,20d后复查头颅MRI示“左侧侧脑室后角周围白质内病灶较前范围减小,考虑脱髓鞘病变可能大”(图1D、E),遗留性格改变、情绪易波动。2011年8月再次出现视物成双,头颅MRI示“双侧侧脑室后角、右额叶、胼胝体压部、脑桥异常信号,部分有不同程度强化”(图1F-I),再次甲泼尼龙冲击治疗2个月后视物成双缓解,遗留计算力减退。2012年1月再次出现发作性肢体抽搐,每次持续数秒钟缓解,每天发作数次,伴有发作性精神行为异常,烦躁、易哭,复查头颅MRI示“双侧侧脑室前角、后角、额叶、胼胝体、脑干多发异常信号”(图1J-L),诊断为“脱髓鞘病变、脑炎后遗症、器质性精神障碍”,给予甲泼尼龙冲击及抗癫痫治疗2个月余,症状缓解。2012年5月再次出现视物成双、视物模糊,经甲泼尼龙冲击治疗后缓解。2012年10月复发视物成双,行头颅MRI示“双侧侧脑室周围白质区、第四脑室左旁多发异常信号”,再次给予甲泼尼龙冲击后症状缓解。2014年12月患者出现右下肢无力,计算力减退,右下肢肌力III-级,右侧查多克征、巴宾斯基征(+),余神经系统体检未见明显异常,复查头颅MRI平扫+强化示“双侧侧脑室旁、基底节、放射冠区、胼胝体膝部、胼胝体压部多发异常强化信号”(图1M),首次化验血和脑脊液抗NMDAR抗体均为(+),并化验前期复发时冻存血或脑脊液标本的抗NMDAR抗体(表1),诊断为“抗NMDAR脑炎”,经激素+免疫球蛋白冲击治疗后症状缓解。2015年5月患者再次出现肢体抽搐、右下肢无力,体格检查:高级智能减退,右下肢肌力Ⅳ级,余神经系统体检及心肺腹部体检未见明显异常,复查头颅MR平扫+强化示“双侧额叶、半卵圆中心及侧脑室旁多发斑片样异常信号影”(图1N),复查血和脑脊液抗NMDAR抗体均为(+),给予免疫球蛋白冲击治疗后症状缓解,并给予硫唑嘌呤口服。患者既往体健,家族中无类似症状者。患者2014年12月诊治过程中先后进行了7次腰椎穿刺检查,脑脊液白细胞数0-24×10^6/L,脑脊液蛋白0.37-0.80g/L,余脑脊液常规、生化、免疫球蛋白、髓鞘碱性蛋白、寡克隆区带电泳、新型隐球菌、抗酸杆菌及细菌、真菌培养及IgG指数均为正常。诊疗过程中多次化验血常规、肝功能、肾功能、生化、甲状腺功能五项、心肌酶、凝血检验、红细胞沉降率、抗O、类风湿因子、C反应蛋白、免疫球蛋白、补体、抗核抗体谱、抗中性粒细胞胞质抗体、肿瘤抗神经系统抗体谱、血肿瘤标志物谱、乙型肝炎病毒、丙型肝炎病毒、人类免疫缺陷病毒、梅毒螺旋体、TORCH十项、血和脑脊液水通道蛋白4(aquaporin-4,AQP4)、抗神经节苷脂、髓鞘少突胶质细胞糖蛋白抗体(myelin oligodendroeyte glycoprotein,MOG)均正常。脑电图示轻中度异常(全头区可见弥漫性θ波)。先后3次视觉诱发电位检查示“双眼分别刺激左、中、右枕记录,视觉诱发电位P100潜伏期均延长”。脊髓、视神经MRI平扫未见明显异常。心电图正常。彩超检查肝、胆、胰、脾、双肾、输尿管、膀胱、前列腺均未见明显异常。胸部CT示两肺纹理增多。2015年7月全身氟代脱氧葡萄糖(FDG)-PET示“双侧半卵圆中心及放射冠区多发斑片状稍低密度灶,FDG代谢未见明显异常,多发性硬化(MS)可能;前列腺腺体两侧FDG代谢异常增高,建议前列腺B超检查及总前列腺特异抗原(tPSA)、游离前列腺特异抗原(fPSA)检查”。复查前列腺B超未见明显异常,复查tPSA、fPSA均正常。
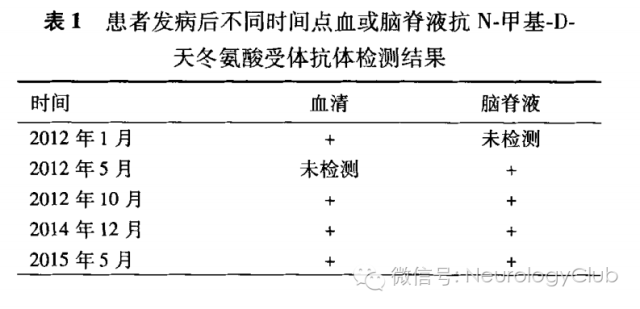
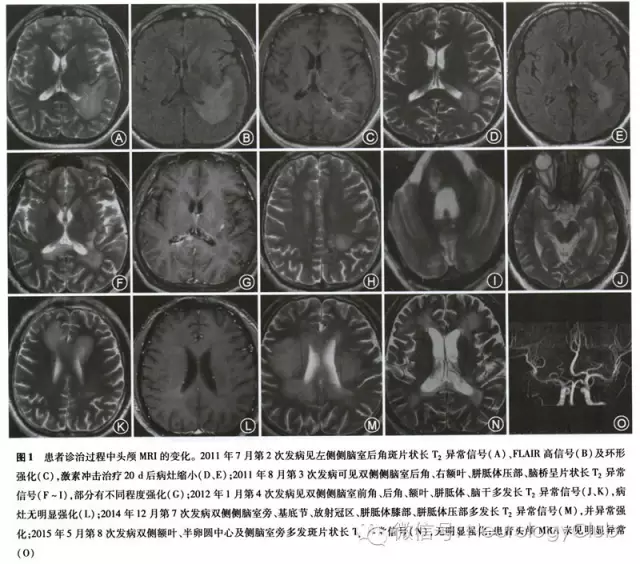
本例患者先后反复发作8次,诊治过程中曾被诊断“病毒性脑炎、中枢神经系统脱髓鞘、MS”等疾病,2014年12月第7次以右下肢偏瘫复发时德国欧蒙间接免疫荧光染色检测血和脑脊液抗NMDAR抗体均为阳性,复查前期3次发作时冻存的血或脑脊液抗NMDAR抗体为阳性,抗NMDAR脑炎具有一次复发和多次复发的可能,因此本例诊断复发性抗NMDAR脑炎明确。本例患者以视物模糊、复视为首发症状,缺乏抗NMDAR脑炎的典型临床特点,误诊为“病毒性脑炎”,病程中有4次以视物模糊和(或)复视复发,这在抗NMDAR脑炎中较少见,而常见于中枢神经系统脱髓鞘疾病;患者头MRI显示双侧侧脑室前后角、额叶、胼胝体、脑桥、基底节、放射冠区、半卵圆中心等多部位受累,白质受累为主,受累部位呈长T2、FLAIR高信号,部分病灶可见强化,每次发作累及部位不同,激素或免疫球蛋白冲击后病灶可缩小或消失,具有时间多发性和空间多发性,仅从影像学上极易误诊为“MS、中枢神经系统炎性脱髓鞘”。该患者癫痫发作、精神行为异常、智能减退等症状在MS中很少出现,临床缺乏MS的典型症状以及反复化验脑脊液寡克隆区带电泳、IgG指数等均为阴性,被误诊为“脑炎后遗症、精神障碍”。尽管病程中多次化验AQP4均为阴性,且脊髓、视神经MRI平扫未见明显异常,但双眼视觉诱发电位P100潜伏期均延长,不能除外存在AQP4阴性的视神经脊髓炎谱系疾病的可能。化验红细胞沉降率、抗链“O”、C反应蛋白、风湿病多肽抗体、抗核抗体谱、抗中性粒细胞胞质抗体等均为阴性,排除了结缔组织相关性脑白质病变;化验乙型肝炎病毒、丙型肝炎病毒、人类免疫缺陷病毒、梅毒螺旋体及TORCH病毒十项均阴性,排除了感染性疾病;化验甲状腺功能五项及甲状腺彩超均正常,排除了甲状腺相关性脑病。随着临床报道病例的逐渐增多,抗NMDAR脑炎既可以与肿瘤伴发,也可以不伴有肿瘤,不伴发肿瘤的抗NMDAR脑炎患者复发率较伴发肿瘤患者要高,国内曾报道3年内复发率达31%;此外,Titulaer等回顾了691例抗NMDAR脑炎患者,发现23例有典型的临床脱髓鞘表现,部分患者AQP4、MOG抗体阳性。抗NMDAR脑炎可以伴有视神经脊髓炎、视神经脊髓炎谱系疾病等中枢神经系统脱髓鞘的发生,也可引起中枢神经系统多发性髓鞘脱失。本患者多次化验血肿瘤标志物、肿瘤抗神经系统抗体谱和彩超、CT检查未见明显异常,全身FDG-PET检查示前列腺腺体两侧FDG代谢异常增高,复查tPSA、fPSA以及前列腺B超正常,目前为止患者还未发现明确存在的肿瘤,仍需定期复查前列腺以排除肿瘤性病变。综上可见,本病例是以多发中枢神经系统脱髓鞘为主要特点、不伴肿瘤的复发性抗NMDAR脑炎,激素冲击、大剂量免疫球蛋白等免疫治疗可有效缓解,其反复发作可能与间歇期没有正规使用免疫抑制剂相关。
总之,抗NMDAR脑炎受累部位多、临床表现多样,容易误诊为或合并MS、视神经脊髓炎、视神经脊髓炎谱系疾病等中枢神经系统脱髓鞘病及病毒性脑炎、脑病、急性精神障碍等疾病,对缺乏典型症状的可疑患者尽早筛查血和脑脊液抗NMDAR抗体尤为重要,明确诊断后即给予激素、免疫球蛋白冲击治疗或血浆置换,筛查是否存在肿瘤,及时诊治预后较好。对于未发现肿瘤的抗NMDAR脑炎患者,在初始免疫治疗结束后,给予维持免疫抑制治疗(吗替麦考酚酯或硫唑嘌呤)至少1年,复发期仍应重视肿瘤的检查。
[参考文献]
胡怀强, 金善, 曹霞, 常高峰, 曹秉振.伴多发性硬化样脱髓鞘的复发性抗N-甲基-D-天冬氨酸受体脑炎一例.中华神经科杂志.2016年4期
神经病学俱乐部 微信号:NeurologyClub神经病学俱乐部,立足一线临床,服务神经科同行,助力神经病学天天向上